
- Title
-
Whole genome sequencing delineates regulatory, copy number, and cryptic splice variants in early onset cardiomyopathy
- Authors
- Lesurf, R., Said, A., Akinrinade, O., Breckpot, J., Delfosse, K., Liu, T., Yao, R., Persad, G., McKenna, F., Noche, R.R., Oliveros, W., Mattioli, K., Shah, S., Miron, A., Yang, Q., Meng, G., Yue, M.C.S., Sung, W.W.L., Thiruvahindrapuram, B., Lougheed, J., Oechslin, E., Mondal, T., Bergin, L., Smythe, J., Jayappa, S., Rao, V.J., Shenthar, J., Dhandapany, P.S., Semsarian, C., Weintraub, R.G., Bagnall, R.D., Ingles, J., Genomics England Research Consortium, Melé, M., Maass, P.G., Ellis, J., Scherer, S.W., Mital, S.
- Source
- Full text @ NPJ Genom Med
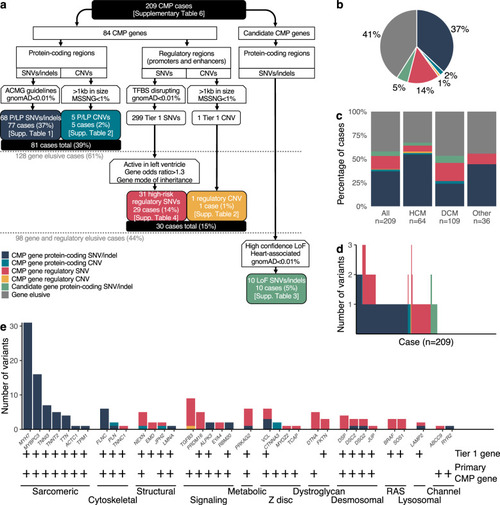
|
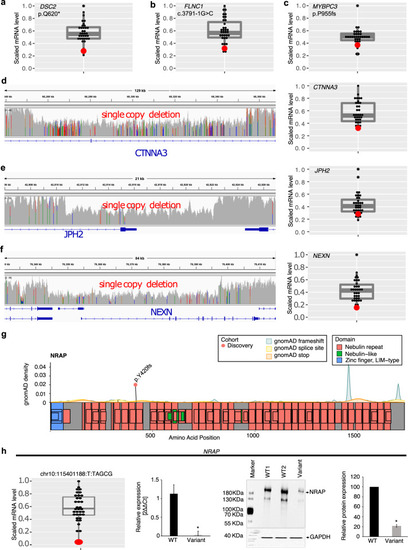
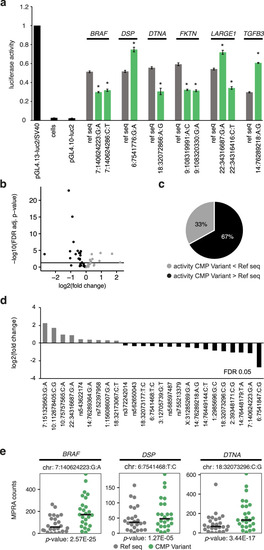
The figure shows LV myocardial gene expression using RNA sequencing in the patient harboring a loss of function or copy number deletion (red dot) compared to other cases without the variant (gray dots) ( |
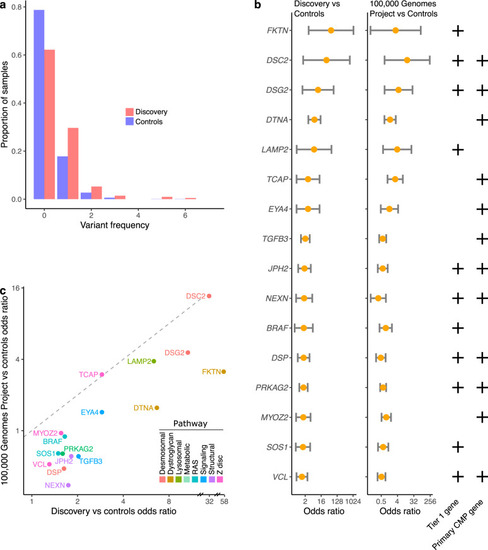
|
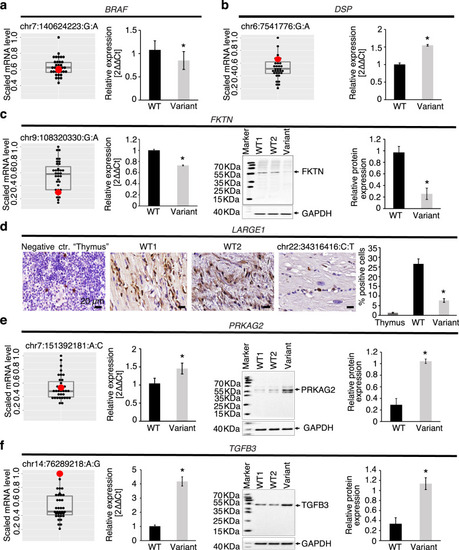
RNA Seq, qRT-PCR, Western blot, and immunohistochemistry were performed in available LV myocardium from CMP patients ( |
|

ZFIN is incorporating published figure images and captions as part of an ongoing project. Figures from some publications have not yet been curated, or are not available for display because of copyright restrictions. PHENOTYPE:
|